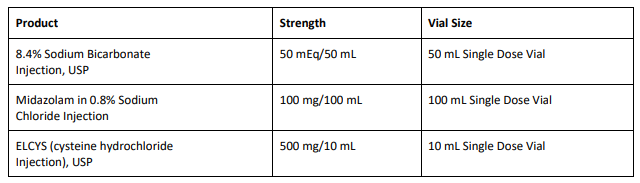
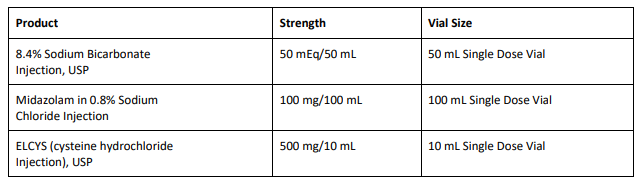
FDA Approves Tyenne (tocilizumab-aazg), a Biosimilar to Actemra
Fresenius Kabi, announced today that the U.S. Food and Drug Administration (FDA) has approved Tyenne® (tocilizumab-aazg), its tocilizumab biosimilar referencing Actemra® (tocilizumab). Tyenne becomes the first tocilizumab biosimilar with both IV and subcutaneous formulations approved by the FDA. In accordance with its patent settlement agreement with Genentech, Fresenius Kabi has a license to market its tocilizumab products in the U.S. commencing on the license dates, which are confidential.
Tyenne has launched globally in more than 10 countries, with plans to launch in many more countries between 2024 and 2025. Tyenne (tocilizumab-aazg) is FDA approved for the treatment of several inflammatory and immune diseases, including rheumatoid arthritis, giant cell arteritis, polyarticular juvenile idiopathic arthritis, and systemic juvenile idiopathic arthritis.